Replication stress characterizes cancer and is associated with incessant and uncontrolled cell replication [61]. Replication stress arises when replication forks are hindered by DNA abnormalities such as DNA lesions [62], R-loops, and secondary DNA structures. The replication stress response represents a deviation from a normal DNA replication program in rapidly dividing cancer cells with poor quality control during replication. In response to various DNA replication problems, activation of ATR kinase and its downstream effector kinase CHK1 is responsible for the replication stress response [63, 64]. ATR phosphorylates checkpoint kinases 1 and 2 (CHK1 and CHK2) to stall the replication cycle when single-stranded DNA breaks mount, giving cells time to repair their DNA. There are several sources of replication stress as discussed below.
R-loops
R-loops are three-stranded hybrid nucleic acid structures formed during a normal transcription process (Fig. 2). Physiologically, R-loops are involved in transcription initiation-termination, Ig class switch recombination, and telomerase elongation [65]. When the homeostasis of an R-loop is altered, the progression of replication is stalled by the phenomenon of transcription-replication conflict. During this conflict, machineries of DNA replication and transcription collide [66] leading to replication stress. If the stress is not effectively resolved in a timely manner, persistence of R-loops can become the source of replication stress and subsequent genomic instability [67, 68]. The presence of a persistent R-loop causes activation of the ATR pathway through an as yet unknown mechanism [69, 70]. Aberrant R-loops that fail to be resolved interfere with DNA damage repair and transcription [71] (reviewed in [68]). Recently, loss of ARID1A has been reported to lead to replication stress and R-loop formation [72]. ARID1A loss generates replication stress through DNA topoisomerase (TOP2A) mis-localization in the genome. By modulating DNA topology, TOP2A is involved in chromosome condensation, separation of chromatids, DNA replication, and transcription [73, 74]. Thus, ATR activation is critical for mitigating R-loop aberrations, and the ATR pathway is responsible for protecting the genome by suppressing transcription-replication conflicts, promoting replication-fork recovery, and arresting cells at G2-M phase for further repair. These new data suggest that ATR inhibition may preferentially kill ARID1A-deficient tumors.
DNA catenation
Another source of replication stress is DNA catenation, an intermediate product of DNA replication. It is formed when two replication forks collide and DNA topoisomerase II removes these structures (known as decatenation) [75]. Mechanistically, TOP2A-mediated DNA decatenation requires BAF complex subunits such as BRG1 (SMARC4) and BAF250A (ARID1A) to bind to chromatin. In the absence of ARID1A, TOP2A is unable to complete this process before mitosis, and as a result, complex chromosomal structures can arise during DNA replication. Moreover, ARID1A loss leads to DNA decatenation defects in tumor cells, which is thought to be due to improper localization of TOP2A [33, 73]. TOP2A helps resolve topological issues brought on by chromosomal metabolism, such as catenated DNA and transcription-replication conflicts. It appears that the failure of TOP2A to properly localize in concert with delayed cell cycle progression in ARID1A deficient cells would result in an increased requirement for ATR function, suggesting a therapeutic potential of ATR inhibitor.
Apurinic/apyrimidinic site
Apurinic/apyrimidinic sites (AP sites), also known as abasic sites, are DNA repair intermediates that act as roadblocks to replication fork progression [61]. These lesions are generally repaired, but replicative DNA polymerases using the Translesion Synthesis (TLS) pathway can bypass these lesions if they are not resolved in a timely fashion [76]. Alkylating drugs like temozolomide and mitomycin-C methylate DNA bases and those methylated bases are repaired by both base excision repair (BER) and nucleotide excision repair (NER) [77]. Recently, we have observed that that ARID1A-deficient cancer cells challenged with temozolomide and PARP inhibitor exhibited significant replication stress, replication fork instability, and DNA damage due to impaired BER arising from the ARID1A deficiency, leading to cell death (Fig. 2). We showed that ARID1A-deficient cancer cells developed a high susceptibility to PARP inhibitors in the presence of TMZ and link BER and TLS functionally. Loss of ARID1A delayed arrival of repair proteins at the AP sites compromising BER. Compromised BER and TLS may exacerbate replication fork stalling in ARID1A-deficient cells, resulting in fork collapse and cell death. Moreover, our preclinical study warrants further clinical testing for the FDA-approved alkylating agent TMZ and PARPi combination, which exhibits synergistic benefits in the ARID1A-deficient tumors (manuscript submitted).
The identification of the underlying genetic alterations across all major subtypes of cancer has provided unprecedented insight into DNA repair proteins that are altered in cancer (26) and has laid the foundation for several precision medicine approaches. Almost every tumor has at least one gene in the replication stress response altered, providing ample opportunities to exploit tumor-specific alterations without affecting normal cells. The concept of synthetic lethality (73), whereby the presence of cancer-specific mutations in specific genes renders other genes essential for cell proliferation and survival, has been fundamental for the discovery of several of these gene-targeted strategies (74). Perhaps the best example of this strategy is the successful use of PARP inhibitors for the treatment of BRCA1/BRCA2-deficient breast and ovarian cancers, as these double-strand break repair defective tumors are dependent on PARP activity for cell survival (37, 75). Considerable effort is now being expended to identify additional synthetic lethal interactions with DNA repair genes, including replication stress response genes. One of the most promising targets currently being explored is ATR. Tumors harboring mutations in genes encoding the chromatin remodeler ARID1A or the DNA damage kinase ATM are synthetically sensitive to ATR inhibitors, with a complete response observed in a patient with metastatic ATM-deficient colorectal cancer (76–78). The dependency of ARID1A-deficient tumors on ATR has been attributed to the mitotic defects present in these cells, which causes an increased reliance on ATR function. Both ARID1A and ATM are frequently altered in tumors, particularly in ovarian and endometrial cancers for ARID1A and lymphoid malignancies for ATM, illustrating the applicability of these findings. In addition to further interactions with ATR, tumors containing mutations in other frequently altered replication stress response genes including RPA1 (NCT03207347 and NCT03377556) and the mutagenic DNA polymerase POLQ (79) are currently being explored to identify druggable synthetic lethal interactions. Genetic interactions with replication stress–inducing agents including CHK1 (80, 81) and WEE1 (82) inhibitors are also being investigated for use in mutation-specific tumor backgrounds, such as the use of CHK1 inhibitors in EZH2-deficient T-cell acute lymphoblastic leukemias.
DNA double-strand breaks (DSBs) are highly deleterious, with a single unrepaired DSB being sufficient to trigger cell death. Compared to healthy cells, cancer cells have a higher DSB burden due to oncogene-induced replication stress and acquired defects in DNA damage response (DDR) mechanisms. Consequently, hyperproliferating cancer cells rely on efficient DSB repair for their survival. Moreover, augmented DSB repair capacity is a major cause of radio- and chemoresistance and, ultimately, cancer recurrence. Although inherited DDR defects can predispose individuals to develop certain cancers, the very same vulnerability may be therapeutically exploited to preferentially kill tumor cells. A paradigm for DNA repair targeted therapy has emerged in cancers that exhibit mutations in BRCA1 or BRCA2 tumor suppressor genes, conferring a strong defect in homologous recombination, a major and error-free DSB repair pathway. Clinical validation of such approaches, commonly described as synthetic lethality (SL), has been provided by the regulatory approval of poly(ADP-ribose) polymerase 1 inhibitors (PARPi) as monotherapy for BRCA1/2 -mutated breast and ovarian tumors. In this review, we will describe the different DSB repair mechanisms and discuss how their specific features could be exploited for cancer therapy. A major emphasis is put on advances in combinatorial treatment modalities and SL approaches arising from DSB repair pathway interdependencies.
Introduction
The integrity of our genome is constantly challenged by endogenous and exogenous insults that can induce DNA damage. To counteract genotoxic threats, cells are equipped with a diverse set of DNA damage signaling and repair mechanisms, collectively known as the DNA damage response (DDR) (1). During tumorigenesis, however, precancerous cells frequently acquire loss-of-function alterations in DDR genes, including core components of selected DNA repair pathways, to accelerate mutagenesis and become malignant (2). While healthy cells have to deal with a minor amount of damage and take advantage of the full DNA repair capacity, malignant cells are frequently equipped with reduced DNA repair functionality to cope with increased replication stress and elevated levels of endogenous DNA damage (3). Consequently, cancer cells become even more dependent on DNA repair mechanisms to survive and proliferate. Conventional treatment modalities such as radiation therapy and certain forms of chemotherapy have been built on the premise to force DNA damage-induced cell death. In summary, cancer cells are often compromised in their ability to adequately process DNA damage, which exerts selective pressure to sustain DNA repair through upregulation of mutagenic pathways, ultimately promoting disease progression and therapy resistance (4, 5).
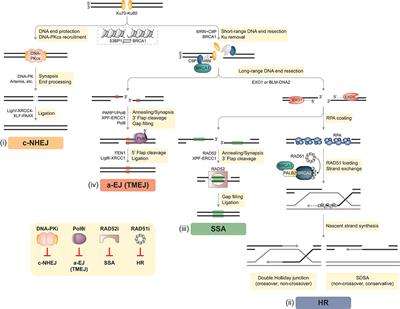
DNA double-strand breaks (DSBs) are considered the most lethal of all DNA lesions, eliciting the majority of the cytotoxic effects induced by ionizing radiation (IR) and certain anti-cancer drugs. Therefore, DSB repair represents a potent and targetable vulnerability in cancer cells. In healthy somatic cells two-ended DSBs are mainly repaired by two pathways: classical non-homologous end joining (c-NHEJ) and homologous recombination (HR) (Figure 1). Auxiliary mechanisms of DSB repair include single-strand annealing (SSA) and alternative end joining (a-EJ) that rely on the presence of larger repeat sequences and microhomologies at the breakpoint, respectively [(6, 7); Figure 1]. Importantly, functional interdependencies between different DNA repair pathways and within compensatory DSB repair mechanisms offer therapeutic opportunities to selectively treat DDR-deficient tumors based on the concept of synthetic lethality (SL) (3, 5, 8, 9).
FIGURE 1
Figure 1. DSB repair pathways in mammalian cells. Two-ended DSBs are preferably repaired by two major competing pathways: classical non-homologous end joining (c-NHEJ) and homologous recombination (HR). In addition, DSBs can be subjected to alternative end joining [a-EJ, also referred to as DNA polymerase theta-mediated end joining (TMEJ)] or single-strand annealing (SSA). BRCA1 and 53BP1 are placed at the center of DSB repair pathway choice. Whereas, chromatin recruitment of 53BP1 drives c-NHEJ, BRCA1 antagonizes 53BP1 to channel DSB repair into HR. (i) C-NHEJ begins with Ku70-Ku80 (Ku) binding to DSB ends, followed by the recruitment of DNA-dependent protein kinase catalytic subunit (DNA-PKcs), forming the DNA-PK holoenzyme implicated in DNA synapsis. If necessary, DNA-PK coordinates limited processing of incompatible or chemically modified DNA ends by nucleases (e.g., Artemis) and other enzymes. The DNA ligase IV (LigIV)-XRCC4-XLF-PAXX complex executes the final ligation step. DNA end resection interferes with the default engagement of c-NHEJ by removing Ku from DNA ends, which is a critical step for initiating HR (ii). First, the MRE11-RAD50-NBS1 (MRN) complex senses the DSB and with the help of BRCA1 and CtIP promotes limited resection of the 5′ strand. Next, more extensive 5′-3′ resection by exonuclease 1 (EXO1), or by the Bloom’s syndrome (BLM) helicase together with the DNA2 nuclease, generates long 3′ ssDNA overhangs that become rapidly coated with the RPA heterotrimer. The BRCA1-PALB2-BRCA2 complex disassembles RAD51 heptamers and loads monomeric RAD51 onto ssDNA, promoting RAD51 filament assembly. Template-dependent strand extension is followed by “synthesis-dependent strand annealing” (SDSA), resulting in a non-crossover gene conversion. Alternatively, capture of the second ssDNA by the D-loop forms a double Holliday junction intermediate, which can be resolved either as a non-crossover or as a crossover. (iii) SSA requires at least 20–25 base pairs (bp) of DNA sequence homology, which are typically found between repetitive elements (indicated as green boxes) in the genome. Subsequently, RAD52 promotes annealing of complementary ssDNA and leftover non-homologous flaps of the 3′ overhangs are cleaved by XPF-ERCC1. The factors that promote gap filling and ligation during SSA remain largely elusive. (iv) In contrast to SSA, a-EJ (or TMEJ) utilizes short microhomologies (MHs) of 2–20 bp (indicated as red boxes) to join the two DNA strands. PARP1 has been implicated in promoting DNA end synapsis and recruiting the specialized DNA polymerase θ (Polθ) to DSBs. Polθ stabilizes MH-mediated joints between the two DNA ends serving as primers for fill-in synthesis. 3′ flaps extending from the joints are removed by XPF/ERCC1. Flap endonuclease 1 (FEN1) has recently been implicated in the removal of 5′ flaps generated by Polθ-mediated strand displacement, while the DNA Ligase III (LigIII)-XRCC1 complex is essential for the final ligation step. Inset, bottom left: DSB repair pathway-specific inhibitors. Inhibition of c-NHEJ has so far been mainly achieved by targeting DNA-PK using different small molecule inhibitors. Strategies to inhibit a-EJ and SSA focus on targeting their respective DNA annealing factors Polθ and RAD52, while the primary target to disrupt HR is RAD51 (see text for more details).
DSB Repair Pathways
The decision as to whether a given DSB is processed by c-NHEJ, HR, or alternative repair pathways is determined by several factors, including genetic and genomic background, DSB complexity, chromatin state, and cell cycle phase. For instance, c-NHEJ operates throughout the cell cycle, whereas HR relies on the presence of an undamaged sister chromatid and is therefore restricted to late S/G2 (7, 10). Therefore, HR activation requires high cyclin-dependent kinase (CDK) activity (11). In addition, numerous HR genes are found upregulated in S/G2 phase of the cell cycle (7). At the chromatin level, the appropriate equilibrium between HR and c-NHEJ is mainly established by BRCA1 and 53BP1, large DDR adaptor proteins that are enriched at DSB sites (12, 13). Whereas, 53BP1 mediates c-NHEJ events and is pivotal in repairing programmed DSBs (e.g., during class-switch recombination), BRCA1 antagonizes 53BP1 to promote DSB resection and HR [(14, 15); Figure 1]. Importantly, one-ended DSBs, predominantly induced by fork breakage or collapse due to high replication stress, lack an adjacent second DNA end for rejoining and can only be repaired by HR-related mechanisms (7).
C-NHEJ
C-NHEJ is accountable for the repair of most two-ended DSBs in mammalian cells (Figure 1). Rapid and high-affinity binding of the Ku70-Ku80 heterodimer (Ku) to DNA ends is followed by the recruitment of DNA-dependent protein kinase catalytic subunit (DNA-PKcs), forming the active DNA-PK holoenzyme. Key functions of DNA-PK in c-NHEJ are (i) promoting synapsis of the broken ends, (ii) coordinating necessary processing of incompatible ends by DNA nucleases (e.g., Artemis) and polymerases, and (iii) engaging the DNA ligase complex composed of DNA ligase IV, XRCC4, XLF, and PAXX (7, 16). Despite rejoining DSBs without the use of extensive sequence homology, c-NHEJ is often highly accurate and its core factors therefore considered as genome “caretakers” (10, 17, 18).
HR
In case c-NHEJ fails or is inappropriate, DSBs are subjected to extensive 5′-end resection, generating 3′-single-stranded (ss) DNA overhangs that interfere with Ku loading and promote high-fidelity repair by HR [(7, 19); Figure 1]. In a first step, the MRE11-RAD50-NBS1 (MRN) complex in conjunction with CtIP, also known as RBBP8, coordinates tethering and short-range nucleolytic degradation of DSB ends (20, 21). MRE11 exhibits a dual endo- and exonuclease activity that is critical for DNA end resection (22). Following long-range resection carried out by EXO1 or the BLM-DNA2 ensemble, the 3′ ssDNA tails are coated by the RPA heterotrimer. In the central step of HR, BRCA2 with the help of BRCA1 and PALB2 delivers RAD51 monomers to ssDNA, resulting in RPA removal and RAD51 presynaptic filament formation required for strand invasion and homology search. Interestingly, in G1 phase, BRCA1-PALB2-BRCA2-RAD51 complex formation is impaired by proteasome-mediated degradation of PALB2 (7). Mechanistically, PALB2-interacting protein KEAP1 in complex with cullin-3-RBX1 ubiquitylate PALB2, thereby suppressing PALB2-BRCA1 (23). HR in somatic cells is mostly completed by synthesis-dependent strand annealing (SDSA), generating non-crossovers, although other outcomes are possible (24).
Alternative DSB Repair Pathways
A-EJ is genetically distinct from Ku-dependent c-NHEJ and RAD51-dependent HR and requires the presence of microhomology (MH) regions (2–20 bp), which are exposed following MRN-CtIP-mediated resection [(25, 26); Figure 1]. Importantly, long-range resection impedes a-EJ and favors HR or SSA (27, 28). DNA polymerase theta (Polθ), a low-fidelity DNA polymerase-helicase, has been recently identified as key factor driving a-EJ by limiting RAD51 nucleation onto ssDNA (29–31). The Polθ-helicase domain displaces RPA from ssDNA tails, whereas the Polθ-polymerase domain promotes their synapsis, thereby facilitating MH-mediated annealing and subsequent gap filling (32, 33). The essential ligation step during a-EJ is performed by the DNA ligase IIIα-XRCC1 complex (26). Contrary to a-EJ, SSA requires more extensive DNA end resection followed by RAD52-mediated annealing of homologous tandem repeat sequences (>20 bp) [(34); Figure 1]. Whether a-EJ and SSA serve primarily as backup pathways in mammalian cells deficient in either c-NHEJ or HR, or are favored at specific genomic loci still remains to be established (35).
DSB Repair Protein Dysfunction in Cancer
Only a minor number of human cancers are associated with downregulation or alterations of core c-NHEJ genes (36). Rare mutations in LIG4 (encoding DNA ligase IV), XLF, DCLRE1C (encoding Artemis) or PRKDC (encoding DNA-PKcs) have been identified in a radiosensitive sub-class of patients with severe combined immunodeficiency (SCID) and can predispose to cancer (37, 38). As c-NHEJ is the predominant DSB repair pathway in human cells, complete loss-of-function is likely to drive cell death due to an unreasonably high DSB burden (36). Elevated DNA-PKcs levels were implicated in the progression of various types of tumors such as prostate cancer and melanoma (36). Noteworthy, PRKDC is with 2.1% the sixth most frequently mutated DNA repair gene in all cancers and considered a potential oncogene, exhibiting frequent copy number gains (39).
A comprehensive analysis of somatic DDR gene alterations delineates HR as the most frequently altered DNA repair pathway across 33 cancer types, most notably ovarian cancer (40). Mutational signatures associated with robust HR deficiency (HRD) primarily included alterations affecting BRCA1, BRCA2, two canonical RAD51 paralog genes (RAD51B, RAD51C), BLM, and RAD50 (40). Large-scale molecular profiling of solid tumor samples across 21 cancer lineages detected pathogenic HR gene mutations with an overall frequency of 17.4%. Here, again, BRCA2 (3%) and BRCA1 (2.8%) were the most commonly mutated bona fide HR genes and predominantly seen in ovarian and breast cancers (41). Heterozygous germline mutations in BRCA1 and BRCA2 are responsible for the majority of hereditary breast and ovarian cancer (HBOC) syndrome patients. However, only ~20–25% of HBOC families have BRCA mutations and other low-to-moderate penetrance HBOC susceptibility genes involved in HR have been identified, including BRIP1, RAD51C, and PALB2 (42). Moreover, revisiting whole-exome sequencing datasets of non-BRCA1/2 familial breast cancer patients confirmed the existence of likely pathogenic germline variants in MRE11A, RAD50, and NBN, encoding components of the MRN complex (43, 44). Lord and Ashworth have coined the term “BRCAness” to denote HRD tumors that share molecular features of BRCA1/2-mutant tumors and are therefore expected to effectively respond to the same treatment modalities (45). Remarkably, however, a recent study indicated that most somatic BRCA1/2 alterations in non-BRCA associated cancer types may be incidental findings unrelated to tumor pathogenesis, rendering them therapeutically irrelevant (46). In contrast to the situation encountered for BRCA1/2, no inactivating mutations of RAD51 have been reported in tumors. Paradoxically, RAD51 is frequently found overexpressed and has been associated with poor prognosis in patients with solid malignancies, thus potentially acting as a driver of aberrant HR (47).
Similarly, elevated MRN expression has been correlated with tumor progression and poor survival in patients with rectal and gastric carcinomas and prostate cancer (48–50). However, with the exception of a positive relationship between MRN deficiency and microsatellite instable (MSI) colorectal cancers, large scale studies will be required to substantiate its relevance in clinical settings (51). Like MRN, CtIP also has rather oncogenic potential at the cellular level, most likely by facilitating a-EJ-dependent chromosomal instability (52–54). Accordingly, mice heterozygous for a null Ctip allele did not display increased tumor susceptibility, meanwhile CtIP inactivation suppressed mammary tumorigenesis caused by p53 deficiency (55). Although still far from being fully characterized, a-EJ is intrinsically mutagenic, typically generating deletions at the repair junction, and suggested to be a major driving force of genomic instability in human cancers (56–58). In particular, a-EJ reliant on Polθ, also referred to as theta mediated end joining (TMEJ, see Figure 1), has emerged as a distinct DSB repair pathway acting predominantly in HRD tumors or on breaks incompatible with c-NHEJ and HR (59). Consistently, depletion of BRCA1/2 resulted in increased usage of TMEJ using reporter assays in human cells (25). Elevated POLQ (encoding for Polθ) expression has been described in numerous cancer types, including breast and ovarian cancer (29, 59–61). Overall, CtIP and Polθ may drive tumorigenesis through a-EJ in defined biological contexts and therefore represent promising therapeutic targets.
DSB Repair Proteins as Drug Targets
As outlined above, DSB repair constitutes an Achilles’ heel of cancer cells and there is a continuous search for compounds specifically targeting DSB repair components to exploit this key vulnerability.
Combinatorial Treatment Regimens Involving DSB Repair Inhibitors
DNA repair targeted therapy was first considered most beneficial in combination with conventional DNA-damaging agents (62, 63). In recent years, mainly thanks to the development of PARPi, additional treatment strategies including DDR inhibitor combinations have been implemented in clinical trials (3, 64, 65). Furthermore, DDR-targeting drugs were found to enhance the effectiveness of immunotherapy by fostering increased immunogenic surveillance and restricted tumor growth (66, 67). An elevated mutation load was shown to increase neoantigen levels in cancer cells thereby promoting tumor immunogenicity (68). Here, we will mainly focus on available DSB repair pathway inhibitors and their synergistic effect in combination with standard chemo- or radiotherapy. Moreover, existing PARPi-based combination strategies will also be highlighted.
Pharmacological Targeting of c-NHEJ
Restraining c-NHEJ capacity has been primarily achieved by targeting DNA-PK (Figure 1). Conceptually, compounds blocking c-NHEJ are thought of as being most effective when used in combination with radiation therapy, as c-NHEJ is taking care of roughly 80% of IR-induced DSBs (69). Whereas, numerous DNA-PKcs small-molecule inhibitors (DNA-PKi) have been developed over the last 20 years, only one specific agent is known to target the Ku heterodimer (70). Weterings et al. identified a compound interfering with the binding of Ku to DNA and sensitizing human cell lines to IR (71). Similarly, the majority of DNA-PKi displayed synergistic effects with IR and chemotherapeutics including etoposide and cisplatin (72). For example, VX-984 induced radiosensitivity of glioblastoma cells grown as orthotopic xenografts (73), whereas combination of the DNA-PKi KU-0060648 with ATR inhibitor AZD6738 potentiated radiosensitization of head and neck squamous cell carcinoma cell lines (74). The most potent DNA-PKi (M3814, CC-115 and CC-122) are currently being investigated in several clinical trials (72). Of particular interest, a dose escalation phase I clinical trial combines M3814 with Avelumab (NCT03724890), a human monoclonal antibody targeting the protein programmed death-ligand 1 (PD-L1). Remarkably, CC-115, a dual inhibitor targeting DNA-PK and the structurally related mammalian target of rapamycin kinase (TORK), was shown to induce caspase-dependent cell death in primary chronic lymphocytic leukemia (CLL) cells and to be clinically effective in CLL patients with an ATM mutation (75, 76). However, it remains an open question of whether DNA-PKi act solely by impairing DSB repair, as other cellular functions of DNA-PKcs have been reported, including cell cycle progression, transcription and telomere maintenance (70).
Pharmacological Targeting of HR
MRE11 harbors endo- and exonuclease activity essential for DNA end resection, thereby channeling DSBs into homology-directed repair pathways (22). A forward chemical genetic screen identified mirin as the first MRE11 inhibitor targeting its exonuclease activity and preventing ATM activation (77). In addition, structure-guided nuclease-specific MRE11 inhibitors revealed that endonuclease inhibition promotes c-NHEJ in lieu of HR, whereas exonuclease inhibition caused a more profound DSB repair defect (78, 79). CtIP’s role in DSB resection has been mostly attributed to its interaction with and stimulation of MRE11, although intrinsic CtIP endonuclease activities have also been demonstrated (80–83). Intriguingly, CtIP-specific inhibitors have not been reported yet. However, inhibition of Bromodomain-containing protein 4 (BRD4) was found to induce an HRD signature by decreasing transcriptional activity of the CtIP promoter and enhancer (84). Reduced CtIP protein levels correlated with increased PARPi sensitivity, potentially qualifying CtIP as a predictive marker for PARPi response. Consistently, different BRD4 inhibitors (e.g., JQ1 and AZD5153) sensitized a broad range of tumor types to PARPi in multiple in vitro and in vivo models (85).
BRCA1 and BRCA2 represent challenging targets for structure-based drug discovery, as they are both large proteins made up of short, functional domains, serving as hubs for multiple protein-protein interactions, interspersed by long, intrinsically disordered linkers (86). In this regard, Pessetto et al. identified a cell permeable peptide ablating phosphoprotein binding by the BRCA1 tandem BRCT domains and enhancing PARPi sensitivity of cancer cells (87). Similarly, a BRCA2-mimetic cell-penetrating peptide disrupting BRCA2-RAD51 interaction conferred PARPi sensitivity in cancer cell lines (88). Small molecules selectively targeting BRCA1’s ubiquitin ligase activity, which is mediated by the N-terminal RING domain and required for efficient DSB resection (89), might also offer a valid alternative to inhibit HR.
Chemical inhibitors of RAD51 (e.g., B02, IBR2, RI-1/2) have been reported to either interfere with RAD51 oligomerization, filament formation or DNA binding, and, ultimately, to induce HR deficiency [(78, 90–94); Figure 1]. Triple combination of B02, the PARPi veliparib and a p38 MAPkinase inhibitor (LY2228820) significantly reduced primary tumor growth in an orthotopic triple negative breast cancer (TNBC) mammary xenograft model (95). Similarly, cancer cell proliferation in a breast cancer xenograft model and in a chronic myelogenous leukemia model bearing the BCR-ABLT315I mutation was significantly slowed upon IBR2 treatment (94). RI-1 potentiated the effect of the alkylating agent Iomustine on a glioma xenograft model, reduced growth of cervical cancer xenografts and hindered TNBC growth in vivo when combined with veliparib (96–98). Based on these preclinical findings, RAD51i were proposed as potential candidates for a novel class of broad-spectrum therapeutics for difficult-to-treat cancers. Interestingly, Cyteir Therapeutics is currently recruiting patients for a phase 1/2 study with CYT-0851, an oral RAD51i designed to reduce the ability of RAD51 to migrate to and from sites of excessive DNA damage (NCT03997968). In addition to direct RAD51 inhibition, inactivation of RAD51 can also be achieved by indirect mechanisms, including tyrosine kinase inhibitors (93). For example, it was recently reported that cediranib (AZD-2171), a potent inhibitor of vascular endothelial growth factor (VEGF) tyrosine kinases, constrains HR through transcriptional repression of RAD51 and BRCA1/2 (99). Accordingly, combination of the PARPi olaparib with cediranib showed superior progression-free and overall survival outcomes in relapsed ovarian cancer patients without documented BRCA1/2 mutations (100).
Even though drugs inhibiting c-NHEJ or HR have proven highly effective in combinatorial treatment strategies, they usually lack tumor specificity and receiving patients often suffer from toxic side effects, resulting in a narrow therapeutic window. Nowadays, SL-based strategies provide a more promising approach for therapeutic interventions, particularly in patients with HRD.
Exploiting Synthetic Lethality in HR-Defective Tumors
The most popular synthetic lethal interaction (SLI) exploited in cancer therapy is the one between BRCA and PARP1 genes (101, 102). Catalytic inhibition of PARP1 “traps” PARP1 molecules on damaged DNA, resulting in replication fork collapse and DSB formation. In combination with HRD, due to BRCA1/2 loss, PARP trapping leads to persistent accumulation of DSBs, inducing cell cycle arrest and apoptosis (Figure 2A). In two landmark studies, pharmacological targeting of PARP1 with the orally active PARPi olaparib showed a favorable therapeutic index in homozygous BRCA-mutated breast or ovarian cancer (103, 104). There are currently six small-molecule PARPi available in the clinic, four of them (olaparib, rucaparib, niraparib and talazoparib) have already obtained approval in different therapeutic settings (65). Despite this remarkable success story, resistance to PARPi remains a major problem in the clinic and an active area of research (105). Nonetheless, the identification of additional, cancer-specific SL gene pairs holds great promise in developing effective monotherapy regimens, as exemplified below (Figure 2B).
FIGURE 2
Figure 2. Synthetic lethal (SL) strategies to target HR-deficient (HRD) tumors. (A) PARPi (red dot) trap PARP1 proteins on endogenous DNA lesions, including single-strand breaks or gaps. If not removed timely, trapped PARP1 blocks the replication machinery, leading to one-ended DSBs that need to be repaired by HR. In HRD tumors such as BRCA1– or BRCA2-mutated cancers, DSBs persist and accumulate, ultimately causing cell death due to SL. (B) HRD tumors are addicted to alternative, mutagenic DSB repair process (a-EJ and SSA) to sustain proliferation. Targeting Polθ (or FEN1) or RAD52 by small molecule inhibitors (red dots) would eliminate a-EJ or SSA, respectively, resulting in SL. (C) Apurinic/apyrimidinic (AP) sites (black dot) are one of the most frequent spontaneous lesions in DNA. If not timely removed by APE2, they have the potential to block DNA replication. Consequently, APE2 inhibition (red dot) would lead to massive accumulation of AP sites associated with increased rates of fork collapse and DSB formation. In healthy cells, HR can deal with those DSBs, promoting cell survival. In contrast, treating HRD tumors with APE2i would cause DSB-induced cell death due to SL.
Two seminal studies from the Sfeir and D’Andrea laboratories established that HRD cancers display a pronounced dependency on TMEJ to limit the toxicity of DSBs [(29, 30); Figure 2B]. Moreover, the fact that Polθ is generally absent in normal cells but upregulated in many cancers makes it a highly desirable drug target (29). Consequently, two established precision oncology companies, Artios Pharma and Repare Therapeutics, have launched Polθ inhibitor programs with first-in-human clinical studies due to start soon. Furthermore, CRISPR-based genetic screens targeting 309 murine DDR genes identified 140 Polq SL genes, including many HR mediators, several c-NHEJ genes and key components of the 53BP1 anti-resection pathway (106). Notably, 30% of human breast cancers in the TCGA cohort were found to be likely deficient in one or more of the 140 Polq SL genes, significantly broadening the number of patients that may benefit from Polθ inhibition (106).
Another interesting SLI was repeatedly reported between RAD52 and BRCA1/2 [(107–111); Figure 2B]. Due to the multiple roles of RAD52 in genome maintenance pathways, the exact mechanism underlying the RAD52-BRCA SL remains to be fully understood (112). However, it has been reported that RAD52-dependent SSA acts as an important backup when direct protein-protein interactions in the BRCA1-PALB2-BRCA2 complex, required to channel resected DSBs down the HR path, are disrupted (113). In large agreement with this notion, RAD52 inhibitors exerted synergistic activity with PARPi against BRCA1-deficient tumor cells (114). Remarkably, combined disruption of RAD52 and POLQ caused additive hypersensitivity to cisplatin, indicating distinct back-up roles in DSB repair and a potentially effective approach for SL therapeutic strategies (115). Several small-molecule RAD52 inhibitors have been developed, but none of them have been subjected to clinical trials (78).
Last but not least, genetic screens by the Elledge laboratory uncovered FEN1 (encoding Flap endonuclease 1) and APEX2 (encoding AP endonuclease 2, APE2) as SL genes in BRCA1/2-deficient backgrounds (116). They proposed that in the context of HRD, FEN1 may be responsible for the removal of Polθ-dependent 5′ flaps during TMEJ (Figures 1, 2B), while APE2 is mainly processing abasic sites at replication forks to avoid fork collapse and DSB formation [(116); Figure 2C].
Notably, acquired genomic instability due to HRD facilitates acquisition of mutations that could trigger therapy resistance (4). For instance, PARPi resistance mechanisms have mostly been linked to either reactivating BRCA mutations or DDR rewiring, thereby functionally restoring HR. In these cases, chemical inhibition of the reactivated HR pathway has been proposed to overcome PARPi resistance (117). Interestingly, numerous studies revealed that reversion mutations of BRCA genes display MH signatures that likely originate from error-prone DSB repair mechanisms such as a-EJ and SSA (118). Consequently, combined inhibition of PARP1 and Polθ (or RAD52) should prolong drug responses and prevent resistance acquisition (118). In addition, targeting alternative SLIs with HRD (Figure 2C) could be beneficial when PARPi resistance arises due to loss of PARP1 expression or activation (117).
Finally, it remains to be said that only few robust SLIs have been identified since the discovery of the SL between PARP inhibition and BRCA1/2 loss of function in 2005 (119). Moreover, it has been argued that most SLIs display incomplete penetrance due to extensive molecular heterogeneity seen in tumors (120). Therefore, assessing the penetrance of SLIs will become an important aspect of future research.
Conclusions
It has become increasingly evident that targeted inhibition of DSB repair proteins offers a wide range of possible applications in cancer treatment. Initially, combinatorial therapy of DSB repair inhibitors with DNA-damaging agents (e.g., IR or cisplatin) were considered most effective. Given that DSB repair deficiency results in increased tumor immunogenicity, the combination of selected DSB repair inhibitors with immunotherapy will very likely find its way into the clinic. In addition, the emerging concept of exploiting SL as anti-cancer therapy is expected to allow more selective and efficient tumor killing without the side-effects of conventional drugs. Importantly, sequential therapy with DNA repair inhibitors was found to be less toxic compared to simultaneous drug administration meanwhile retaining treatment efficacy (121, 122). Consequently, detailed evaluation of the drug administration timing is of vital interest to reduce cytotoxicity. In addition, the stratification of robust biomarkers and detection of mutational signatures will be highly critical to the implementation of SL but also combinatorial therapy regimens (123). Finally, DSBs are repaired by multifactorial pathways that are heavily connected. These interdependencies generate potentially druggable vulnerabilities but also opportunities for tumors to develop drug resistance. Thus, establishing potent inhibitors for each DSB repair pathway will create new treatment opportunities for a wide range of tumors.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
Our research was supported by the Swiss National Science Foundation (grant number: 31003A_176161, to AS), the Swiss Cancer League (grant numbers: KFS-3845-02-2016-R and KFS-4702-02-2019, to AS), and the Forschungskredit of the University of Zurich (grant number: FK-19-037, to AT).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize to authors whose work could not be cited due to space limitations.
References
2. Dietlein F, Thelen L, Reinhardt HC. Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends Genet. (2014) 30:326–39. doi: 10.1016/j.tig.2014.06.003
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer. (2012) 12:587–98. doi: 10.1038/nrc3342
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Scully R, Panday A, Elango R, Willis NA. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol. (2019) 1–17. doi: 10.1038/s41580-019-0152-0
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Motegi A, Masutani M, Yoshioka K-I, Bessho T. Aberrations in DNA repair pathways in cancer and therapeutic significances. Semin Cancer Biol. (2019) 58:29–46. doi: 10.1016/j.semcancer.2019.02.005
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Krenning L, van den Berg J, Medema RH. Life or death after a break: what determines the choice? Mol Cell. (2019) 76:346–58. doi: 10.1016/j.molcel.2019.08.023
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Bunting SF, Callen E, Wong N, Chen H-T, Polato F, Gunn A, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. (2010) 141:243–54. doi: 10.1016/j.cell.2010.03.012
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Daley JM, Sung P. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol Cell Biol. (2014) 34:1380–8. doi: 10.1128/MCB.01639-13
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Difilippantonio MJ, Zhu J, Chen HT, Meffre E, Nussenzweig MC, Max EE, et al. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature. (2000) 404:510–4. doi: 10.1038/35006670
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Zhao W, Wiese C, Kwon Y, Hromas R, Sung P. The BRCA tumor suppressor network in chromosome damage repair by homologous recombination. Annu Rev Biochem. (2019) 88:221–45. doi: 10.1146/annurev-biochem-013118-111058
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Marini F, Rawal CC, Liberi G, Pellicioli A. Regulation of DNA double strand breaks processing: focus on barriers. Front Mol Biosci. (2019) 6:836–8. doi: 10.3389/fmolb.2019.00055
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Syed A, Tainer JA. The MRE11–RAD50–NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu Rev Biochem. (2018) 87:263–94. doi: 10.1146/annurev-biochem-062917-012415
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Orthwein A, Noordermeer SM, Wilson MD, Landry S, Enchev RI, Sherker A, et al. A mechanism for the suppression of homologous recombination in G1 cells. Nature. (2015) 528:422–6. doi: 10.1038/nature16142
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Ahrabi S, Sarkar S, Pfister SX, Pirovano G, Higgins GS, Porter ACG, et al. A role for human homologous recombination factors in suppressing microhomology-mediated end joining. Nucleic Acids Res. (2016) 44:5743–57. doi: 10.1093/nar/gkw326
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Deng SK, Gibb B, de Almeida MJ, Greene EC, Symington LS. RPA antagonizes microhomology-mediated repair of DNA double-strand breaks. Nat Struct Mol Biol. (2014) 21:405–12. doi: 10.1038/nsmb.2786
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Truong LN, Li Y, Shi LZ, Hwang PYH, He J, Wang H, et al. Microhomology-mediated end joining and homologous recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc Natl Acad Sci USA. (2013) 110:7720–5. doi: 10.1073/pnas.1213431110
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Kent T, Chandramouly G, McDevitt SM, Ozdemir AY, Pomerantz RT. Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase θ. Nat Struct Mol Biol. (2015) 22:230–7. doi: 10.1038/nsmb.2961
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Mateos-Gomez PA, Kent T, Deng SK, McDevitt S, Kashkina E, Hoang TM, et al. The helicase domain of Polθ counteracts RPA to promote alt-NHEJ. Nat Struct Mol Biol. (2017) 24:1116–23. doi: 10.1038/nsmb.3494
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Sishc BJ, Davis AJ. The role of the core non-homologous end joining factors in carcinogenesis and cancer. Cancers. (2017) 9:81–30. doi: 10.3390/cancers9070081
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Chae YK, Anker JF, Carneiro BA, Chandra S, Kaplan J, Kalyan A, et al. Genomic landscape of DNA repair genes in cancer. Oncotarget. (2016) 7:23312–21. doi: 10.18632/oncotarget.8196
PubMed Abstract | CrossRef Full Text | Google Scholar
40. Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, et al. Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Reports. (2018) 23:239–54.e6. doi: 10.1016/j.celrep.2018.03.076
PubMed Abstract | CrossRef Full Text | Google Scholar
41. Heeke AL, Pishvaian MJ, Lynce F, Xiu J, Brody JR, Chen WJ, et al. Prevalence of homologous recombination–related gene mutations across multiple cancer types. JCO Precis Oncol. (2018) 2018:1–13. doi: 10.1200/PO.17.00286
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Nielsen FC, van Overeem Hansen T, Sørensen CS. Hereditary breast and ovarian cancer: new genes in confined pathways. Nat Rev Cancer. (2016) 16:599–612. doi: 10.1038/nrc.2016.72
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Bartkova J, Tommiska J, Oplustilova L, Aaltonen K, Tamminen A, Heikkinen T, et al. Aberrations of the MRE11-RAD50-NBS1 DNA damage sensor complex in human breast cancer: MRE11 as a candidate familial cancer-predisposing gene. Mol Oncol. (2008) 2:296–316. doi: 10.1016/j.molonc.2008.09.007
PubMed Abstract | CrossRef Full Text | Google Scholar
44. Yin J, Wu K, Ma Q, Dong H, Zhu Y, Hu L, et al. Revisiting non-BRCA1/2 familial whole exome sequencing datasets implicates NCK1 as a cancer gene. Front Genet. (2019) 10:527. doi: 10.3389/fgene.2019.00527
PubMed Abstract | CrossRef Full Text | Google Scholar
47. Gachechiladze M, Škarda J, Soltermann A, Joerger M. RAD51 as a potential surrogate marker for DNA repair capacity in solid malignancies. Int J Cancer. (2017) 141:1286–94. doi: 10.1002/ijc.30764
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Ho V, Chung L, Singh A, Lea V, Abubakar A, Lim SH, et al. Overexpression of the MRE11-RAD50-NBS1 (MRN) complex in rectal cancer correlates with poor response to neoadjuvant radiotherapy and prognosis. BMC Cancer. (2018) 18:177–111. doi: 10.1186/s12885-018-4776-9
PubMed Abstract | CrossRef Full Text | Google Scholar
49. Li J, Su T, Yang L, Zhang C, He Y. High expression of MRE11 correlates with poor prognosis in gastric carcinoma. Diagn Pathol. (2019) 14:87–9. doi: 10.1186/s13000-019-0844-y
PubMed Abstract | CrossRef Full Text | Google Scholar
50. Wang J, Xu W-H, Wei Y, Zhu Y, Qin X-J, Zhang H-L, et al. Elevated MRE11 expression associated with progression and poor outcome in prostate cancer. J Cancer. (2019) 10:4333–40. doi: 10.7150/jca.31454
PubMed Abstract | CrossRef Full Text | Google Scholar
51. Situ Y, Chung L, Lee C, Ho V. MRN (MRE11-RAD50-NBS1) Complex in human cancer and prognostic implications in colorectal cancer. IJMS. (2019) 20:816–11. doi: 10.3390/ijms20040816
PubMed Abstract | CrossRef Full Text | Google Scholar
52. Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. (2008) 4:e1000110. doi: 10.1371/journal.pgen.1000110
PubMed Abstract | CrossRef Full Text | Google Scholar
53. Yu X, Wu LC, Bowcock AM, Aronheim A, Baer R. The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J Biol Chem. (1998) 273:25388–92. doi: 10.1074/jbc.273.39.25388
PubMed Abstract | CrossRef Full Text | Google Scholar
54. Zhang Y, Jasin M. An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat Struct Mol Biol. (2010) 18:80–4. doi: 10.1038/nsmb.1940
PubMed Abstract | CrossRef Full Text | Google Scholar
55. Reczek CR, Shakya R, Miteva Y, Szabolcs M, Ludwig T, Baer R. The DNA resection protein CtIP promotes mammary tumorigenesis. Oncotarget. (2016) 7(22):32172–83. doi: 10.18632/oncotarget.8605
CrossRef Full Text | Google Scholar
57. Stephens PJ, McBride DJ, Lin M-L, Varela I, Pleasance ED, Simpson JT, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. (2009) 462:1005–10. doi: 10.1038/nature08645
PubMed Abstract | CrossRef Full Text | Google Scholar
58. Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. (2011) 144:27–40. doi: 10.1016/j.cell.2010.11.055
PubMed Abstract | CrossRef Full Text | Google Scholar
59. Schimmel J, van Schendel R, den Dunnen JT, Tijsterman M. Templated insertions: a smoking gun for polymerase theta-mediated end joining. Trends Genet. (2019) 35:632–44. doi: 10.1016/j.tig.2019.06.001
PubMed Abstract | CrossRef Full Text | Google Scholar
60. Higgins GS, Harris AL, Prevo R, Helleday T, McKenna WG, Buffa FM. Overexpression of POLQ confers a poor prognosis in early breast cancer patients. Oncotarget. (2010) 1:175–84. doi: 10.18632/oncotarget.124
PubMed Abstract | CrossRef Full Text | Google Scholar
61. Lemee F, Bergoglio V, Fernandez-Vidal A, Machado-Silva A, Pillaire MJ, Bieth A, et al. DNA polymerase θ up-regulation is associated with poor survival in breast cancer, perturbs DNA replication, and promotes genetic instability. Proc Natl Acad Sci USA. (2010) 107:13390–5. doi: 10.1073/pnas.0910759107
PubMed Abstract | CrossRef Full Text | Google Scholar
62. Gavande NS, VanderVere-Carozza PS, Hinshaw HD, Jalal SI, Sears CR, Pawelczak KS, et al. DNA repair targeted therapy: the past or future of cancer treatment? Pharmacol Ther. (2016) 160:65–83. doi: 10.1016/j.pharmthera.2016.02.003
CrossRef Full Text | Google Scholar
68. Pilié PG, Tang C, Mills GB, Yap TA. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol. (2019) 16:81–104. doi: 10.1038/s41571-018-0114-z
PubMed Abstract | CrossRef Full Text | Google Scholar
69. Shibata A, Jeggo P. A historical reflection on our understanding of radiation-induced DNA double strand break repair in somatic mammalian cells; interfacing the past with the present. Int J Radiat Biol. (2019) 95:945–56. doi: 10.1080/09553002.2018.1564083
PubMed Abstract | CrossRef Full Text | Google Scholar
71. Weterings E, Gallegos AC, Dominick LN, Cooke LS, Bartels TN, Vagner J, et al. A novel small molecule inhibitor of the DNA repair protein Ku70/80. DNA Repair. (2016) 43:98–106. doi: 10.1016/j.dnarep.2016.03.014
PubMed Abstract | CrossRef Full Text | Google Scholar
73. Timme CR, Rath BH, O’Neill JW, Camphausen K, Tofilon PJ. The DNA-PK inhibitor VX-984 enhances the radiosensitivity of glioblastoma cells grown in vitro and as orthotopic xenografts. Mol Cancer Ther. (2018) 17:1207–16. doi: 10.1158/1535-7163.MCT-17-1267
PubMed Abstract | CrossRef Full Text | Google Scholar
74. Hafsi H, Dillon MT, Barker HE, Kyula JN, Schick U, Paget JT, et al. Combined ATR and DNA-PK inhibition radiosensitizes tumor cells independently of their p53 status. Front Oncol. (2018) 8:245. doi: 10.3389/fonc.2018.00245
PubMed Abstract | CrossRef Full Text | Google Scholar
75. Mortensen DS, Perrin-Ninkovic SM, Shevlin G, Elsner J, Zhao J, Whitefield B, et al. Optimization of a series of triazole containing mammalian target of rapamycin (mTOR) kinase inhibitors and the discovery of CC-115. J Med Chem. (2015) 58:5599–608. doi: 10.1021/acs.jmedchem.5b00627
PubMed Abstract | CrossRef Full Text | Google Scholar
76. Thijssen R, ter Burg J, Garrick B, van Bochove GGW, Brown JR, Fernandes SM, et al. Dual TORK/DNA-PK inhibition blocks critical signaling pathways in chronic lymphocytic leukemia. Blood. (2016) 128:574–83. doi: 10.1182/blood-2016-02-700328
PubMed Abstract | CrossRef Full Text | Google Scholar
77. Dupré A, Boyer-Chatenet L, Sattler RM, Modi AP, Lee JH, Nicolette ML, et al. A forward chemical genetic screen reveals an inhibitor of the Mre11–Rad50–Nbs1 complex. Nat Chem Biol. (2008) 4:119–25. doi: 10.1038/nchembio.63
PubMed Abstract | CrossRef Full Text | Google Scholar
78. Hengel SR, Spies MA, Spies M. Small-molecule inhibitors targeting DNA repair and DNA repair deficiency in research and cancer therapy. Cell Chem Biol. (2017) 24:1101–19. doi: 10.1016/j.chembiol.2017.08.027
PubMed Abstract | CrossRef Full Text | Google Scholar
79. Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois M-M, et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell. (2014) 53:7–18. doi: 10.1016/j.molcel.2013.11.003
PubMed Abstract | CrossRef Full Text | Google Scholar
80. Anand R, Ranjha L, Cannavo E, Cejka P. Phosphorylated CtIP functions as a co-factor of the MRE11-RAD50-NBS1 endonuclease in DNA end resection. Mol Cell. (2016) 64:940–50. doi: 10.1016/j.molcel.2016.10.017
PubMed Abstract | CrossRef Full Text | Google Scholar
81. Anand R, Jasrotia A, Bundschuh D, Howard SM, Ranjha L, Stucki M, et al. NBS1 promotes the endonuclease activity of the MRE11-RAD50 complex by sensing CtIP phosphorylation. EMBO J. (2019) 38:2585–2516. doi: 10.15252/embj.2018101005
PubMed Abstract | CrossRef Full Text | Google Scholar
82. Makharashvili N, Tubbs AT, Yang SH, Wang H, Barton O, Zhou Y, et al. Catalytic and noncatalytic roles of the CtIP endonuclease in double-strand break end resection. Mol Cell. (2014) 54:1022–33. doi: 10.1016/j.molcel.2014.04.011
PubMed Abstract | CrossRef Full Text | Google Scholar
83. Wang H, Li Y, Truong LN, Shi LZ, Hwang PY-H, He J, et al. CtIP maintains stability at common fragile sites and inverted repeats by end resection-independent endonuclease activity. Mol Cell. (2014) 54:1012–21. doi: 10.1016/j.molcel.2014.04.012
PubMed Abstract | CrossRef Full Text | Google Scholar
84. Sun C, Yin J, Fang Y, Chen J, Jeong KJ, Chen X, et al. BRD4 inhibition is synthetic lethal with PARP inhibitors through the induction of homologous recombination deficiency. Cancer Cell. (2018) 33:401–16.e8. doi: 10.1016/j.ccell.2018.01.019
PubMed Abstract | CrossRef Full Text | Google Scholar
85. Duan Y, Guan Y, Qin W, Zhai X, Yu B, Liu H. Targeting Brd4 for cancer therapy: inhibitors and degraders. Med Chem Commun. (2018) 9:1779–802. doi: 10.1039/C8MD00198G
PubMed Abstract | CrossRef Full Text | Google Scholar
86. Chen C-C, Feng W, Lim PX, Kass EM, Jasin M. Homology-directed repair and the role of BRCA1, BRCA2, and related proteins in genome integrity and cancer. Ann Rev Cancer Biol. (2018) 2:313–36. doi: 10.1146/annurev-cancerbio-030617-050502
PubMed Abstract | CrossRef Full Text | Google Scholar
87. Pessetto ZY, Yan Y, Bessho T, Natarajan A. Inhibition of BRCT(BRCA1)-phosphoprotein interaction enhances the cytotoxic effect of olaparib in breast cancer cells: a proof of concept study for synthetic lethal therapeutic option. Breast Cancer Res Treat. (2012) 134:511–7. doi: 10.1007/s10549-012-2079-4
PubMed Abstract | CrossRef Full Text | Google Scholar
88. Trenner A, Godau J, Sartori AA. A short BRCA2-derived cell-penetrating peptide targets RAD51 function and confers hypersensitivity toward PARP inhibition. Mol Cancer Ther. (2018) 17:1392–1404. doi: 10.1158/1535-7163.MCT-17-1156
PubMed Abstract | CrossRef Full Text | Google Scholar
89. Densham RM, Morris JR. The BRCA1 Ubiquitin ligase function sets a new trend for remodelling in DNA repair. Nucleus. (2017) 8:116–25. doi: 10.1080/19491034.2016.1267092
PubMed Abstract | CrossRef Full Text | Google Scholar
90. Budke B, Kalin JH, Pawlowski M, Zelivianskaia AS, Wu M, Kozikowski AP, et al. An optimized RAD51 inhibitor that disrupts homologous recombination without requiring michael acceptor reactivity. J Med Chem. (2012) 56:254–63. doi: 10.1021/jm301565b
PubMed Abstract | CrossRef Full Text | Google Scholar
91. Budke B, Logan HL, Kalin JH, Zelivianskaia AS, Cameron McGuire W, Miller LL, et al. RI-1: a chemical inhibitor of RAD51 that disrupts homologous recombination in human cells. Nucleic Acids Res. (2012) 40:7347–57. doi: 10.1093/nar/gks353
PubMed Abstract | CrossRef Full Text | Google Scholar
92. Huang F, Motlekar NA, Burgwin CM, Napper AD, Diamond SL, Mazin AV. Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem Biol. (2011) 6:628–35. doi: 10.1021/cb100428c
PubMed Abstract | CrossRef Full Text | Google Scholar
93. Ward A, Khanna KK, Wiegmans AP. Targeting homologous recombination, new pre-clinical and clinical therapeutic combinations inhibiting RAD51. Cancer Treat Rev. (2015) 41:35–45. doi: 10.1016/j.ctrv.2014.10.006
PubMed Abstract | CrossRef Full Text | Google Scholar
94. Zhu J, Zhou L, Wu G, Konig H, Lin X, Li G, et al. A novel small molecule RAD51 inactivator overcomes imatinib-resistance in chronic myeloid leukaemia. EMBO Mol Med. (2013) 5:353–65. doi: 10.1002/emmm.201201760
PubMed Abstract | CrossRef Full Text | Google Scholar
95. Wiegmans AP, Miranda M, Wen SW, Al-Ejeh F, Möller A. RAD51 inhibition in triple negative breast cancer cells is challenged by compensatory survival signaling and requires rational combination therapy. Oncotarget. (2016) 7:60087–100. doi: 10.18632/oncotarget.11065
PubMed Abstract | CrossRef Full Text | Google Scholar
96. Berte N, Piee-Staffa A, Piecha N, Wang M, Borgmann K, Kaina B, et al. Targeting homologous recombination by pharmacological inhibitors enhances the killing response of glioblastoma cells treated with alkylating drugs. Mol Cancer Ther. (2016) 15:2665–78. doi: 10.1158/1535-7163.MCT-16-0176
PubMed Abstract | CrossRef Full Text | Google Scholar
97. Chen Q, Cai D, Li M, Wu X. The homologous recombination protein RAD51 is a promising therapeutic target for cervical carcinoma. Oncol Rep. (2017) 38:767–74. doi: 10.3892/or.2017.5724
PubMed Abstract | CrossRef Full Text | Google Scholar
98. Shi Y, Jin J, Wang X, Ji W, Guan X. DAXX, as a tumor suppressor, impacts DNA damage repair and sensitizes BRCA-proficient TNBC cells to PARP inhibitors. Neoplasia. (2019) 21:533–44. doi: 10.1016/j.neo.2019.04.001
PubMed Abstract | CrossRef Full Text | Google Scholar
99. Kaplan AR, Gueble SE, Liu Y, Oeck S, Kim H, Yun Z, et al. Cediranib suppresses homology-directed DNA repair through down-regulation of BRCA1/2 and RAD51. Sci Transl Med. (2019) 11:eaav4508. doi: 10.1126/scitranslmed.aav4508
PubMed Abstract | CrossRef Full Text | Google Scholar
100. Liu JF, Barry WT, Birrer M, Lee JM, Buckanovich RJ, Fleming GF, et al. Overall survival and updated progression-free survival outcomes in a randomized phase II study of combination cediranib and olaparib versus olaparib in relapsed platinum-sensitive ovarian cancer. Ann Oncol. (2019) 30:551–7. doi: 10.1093/annonc/mdz018
PubMed Abstract | CrossRef Full Text | Google Scholar
101. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. (2005) 434:913–7. doi: 10.1038/nature03443
PubMed Abstract | CrossRef Full Text | Google Scholar
102. Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. (2005) 434:917–20. doi: 10.1038/nature03445
PubMed Abstract | CrossRef Full Text | Google Scholar
103. Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn K, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. (2010) 376:245–51. doi: 10.1016/S0140-6736(10)60893-8
CrossRef Full Text | Google Scholar
104. Tutt A, Robson M, Audeh MW, Weitzerl JN, Friedlander M, Arun B, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. (2010) 376:235–44. doi: 10.1016/S0140-6736(10)60892-6
CrossRef Full Text | Google Scholar
106. Feng W, Simpson DA, Carvajal-Garcia J, Price BA, Kumar RJ, Mose LE, et al. Genetic determinants of cellular addiction to DNA polymerase theta. Nat Commun. (2019) 10:22–13. doi: 10.1038/s41467-019-12234-1
PubMed Abstract | CrossRef Full Text | Google Scholar
107. Cramer-Morales K, Nieborowska-Skorska M, Scheibner K, Padget M, Irvine DA, Sliwinski T, et al. Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile. Blood. (2013) 122:1293–304. doi: 10.1182/blood-2013-05-501072
PubMed Abstract | CrossRef Full Text | Google Scholar
108. Feng Z, Scott SP, Bussen W, Sharma GG, Guo G, Pandita TK, et al. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc Natl Acad Sci USA. (2011) 108:686–91. doi: 10.1073/pnas.1010959107
PubMed Abstract | CrossRef Full Text | Google Scholar
109. Hengel SR, Malacaria E, Folly da Silva Constantino L, Bain FE, Diaz A, Koch BG, et al. Small-molecule inhibitors identify the RAD52-ssDNA interaction as critical for recovery from replication stress and for survival of BRCA2 deficient cells. Elife. (2016) 5:e14740. doi: 10.7554/eLife.14740
PubMed Abstract | CrossRef Full Text | Google Scholar
110. Huang F, Goyal N, Sullivan K, Hanamshet K, Patel M, Mazina OM, et al. Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids Res. (2016) 44:4189–99. doi: 10.1093/nar/gkw087
PubMed Abstract | CrossRef Full Text | Google Scholar
111. Lok BH, Carley AC, Tchang B, Powell SN. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene. (2012) 32:3552–8. doi: 10.1038/onc.2012.391
PubMed Abstract | CrossRef Full Text | Google Scholar
113. Anantha RW, Simhadri S, Foo TK, Maio S, Liu J, Shen Z, et al. Functional and mutational landscapes of BRCA1 for homology-directed repair and therapy resistance. Elife. (2017) 6:e21350. doi: 10.7554/eLife.21350
PubMed Abstract | CrossRef Full Text | Google Scholar
114. Sullivan-Reed K, Bolton-Gillespie E, Dasgupta Y, Langer S, Siciliano M, Nieborowska-Skorska M, et al. Simultaneous targeting of PARP1 and RAD52 triggers dual synthetic lethality in BRCA-deficient tumor cells. Cell Rep. (2018) 23:3127–36. doi: 10.1016/j.celrep.2018.05.034
PubMed Abstract | CrossRef Full Text | Google Scholar
115. Kelso AA, Lopezcolorado FW, Bhargava R, Stark JM. Distinct roles of RAD52 and POLQ in chromosomal break repair and replication stress response. PLoS Genet. (2019) 15:e1008319. doi: 10.1371/journal.pgen.1008319
PubMed Abstract | CrossRef Full Text | Google Scholar
116. Mengwasser KE, Adeyemi RO, Leng Y, Choi MY, Clairmont C, D’Andrea AD, et al. Genetic screens reveal FEN1 and APEX2 as BRCA2 synthetic lethal targets. Mol Cell. (2019) 73:885–99.e6. doi: 10.1016/j.molcel.2018.12.008
PubMed Abstract | CrossRef Full Text | Google Scholar
118. Gogola E, Rottenberg S, Jonkers J. Resistance to PARP inhibitors: lessons from preclinical models of BRCA-associated cancer. Ann Rev Cancer Biol. (2019) 3:235–54. doi: 10.1146/annurev-cancerbio-030617-050232
CrossRef Full Text | Google Scholar
122. Fang Y, McGrail DJ, Sun C, Labrie M, Chen X, Zhang D, et al. Sequential therapy with PARP and WEE1 inhibitors minimizes toxicity while maintaining efficacy. Cancer Cell. (2019) 35:851–67.e7. doi: 10.1016/j.ccell.2019.05.001
PubMed Abstract | CrossRef Full Text | Google Scholar
123. Ma J, Setton J, Lee NY, Riaz N, Powell SN. The therapeutic significance of mutational signatures from DNA repair deficiency in cancer. Nat Commun. (2018) 9:3292. doi: 10.1038/s41467-018-05228-y
PubMed Abstract | CrossRef Full Text | Google Scholar
https://www.youtube.com/watch?v=6gdmwgZdXRE


